What is the 510(k)?
The 510(k) regulatory pathway is a submission process that medical device manufacturers use to get clearance from the FDA to market an eligible product in the U.S. Under the 510(k) pathway, a medical device manufacturer submits a premarket notification to the FDA, which includes information about the device’s intended use, design, and performance characteristics compared to a predicate device.
The goal of the 510(k) submission is to demonstrate that the device is “substantially equivalent” to a device that has already been cleared by the FDA and is in commercial distribution. This is referred to as a “predicate device.” To make this determination, the FDA reviews the 510(k) submission to ensure that the device is equivalent to the predicate and performs as intended.
Once the FDA determines that the device is substantially equivalent to a predicate device, the agency will issue a clearance letter that allows the manufacturer to market the device in the United States. It’s important to note that clearance under the 510(k) pathway does not indicate that the FDA has approved the device but rather that the FDA has determined the device is equivalent to a similar device already on the market.
FDA 510(k) Preparations and Submissions
In order for a medical device to be sold in the U.S. market many of medical devices (most Class 2, some Class 1 and 3) are subject to FDA clearance called 510(k) submission process. A 510(k) is a pre-market submission made to FDA to demonstrate that the device to be marketed is at least as safe and effective, that is substantially equivalent, to an already marketed device in U.S. The 510(k) submission must compar
e the device to at least one legally marketed devices and provide support for their substantial equivalency claims.
As a device manufacturer or distributer you may carry the uncharted scientific material necessary for 510(k) submission. However, FDA Listing Inc. will assist you with 510(k) filling preparation to ensure all required FDA formats and necessary elements are fulfilled and are FDA compliant before submitting the 510(k) on your behalf. Click the start button for 510(k) submission assistance.
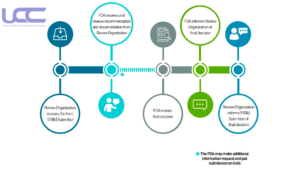
Timeline of Communication during 510(k) Review:
